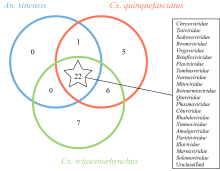
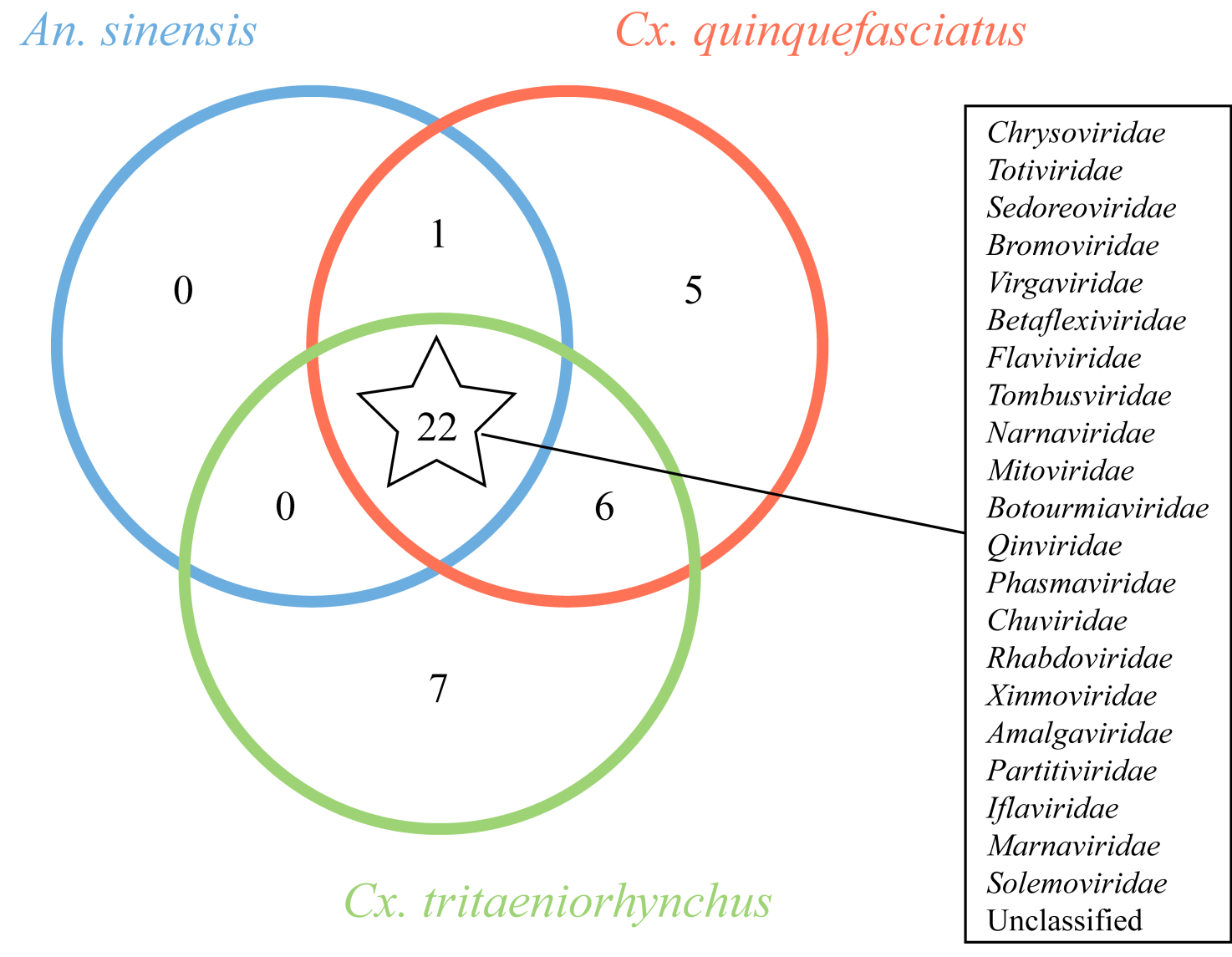
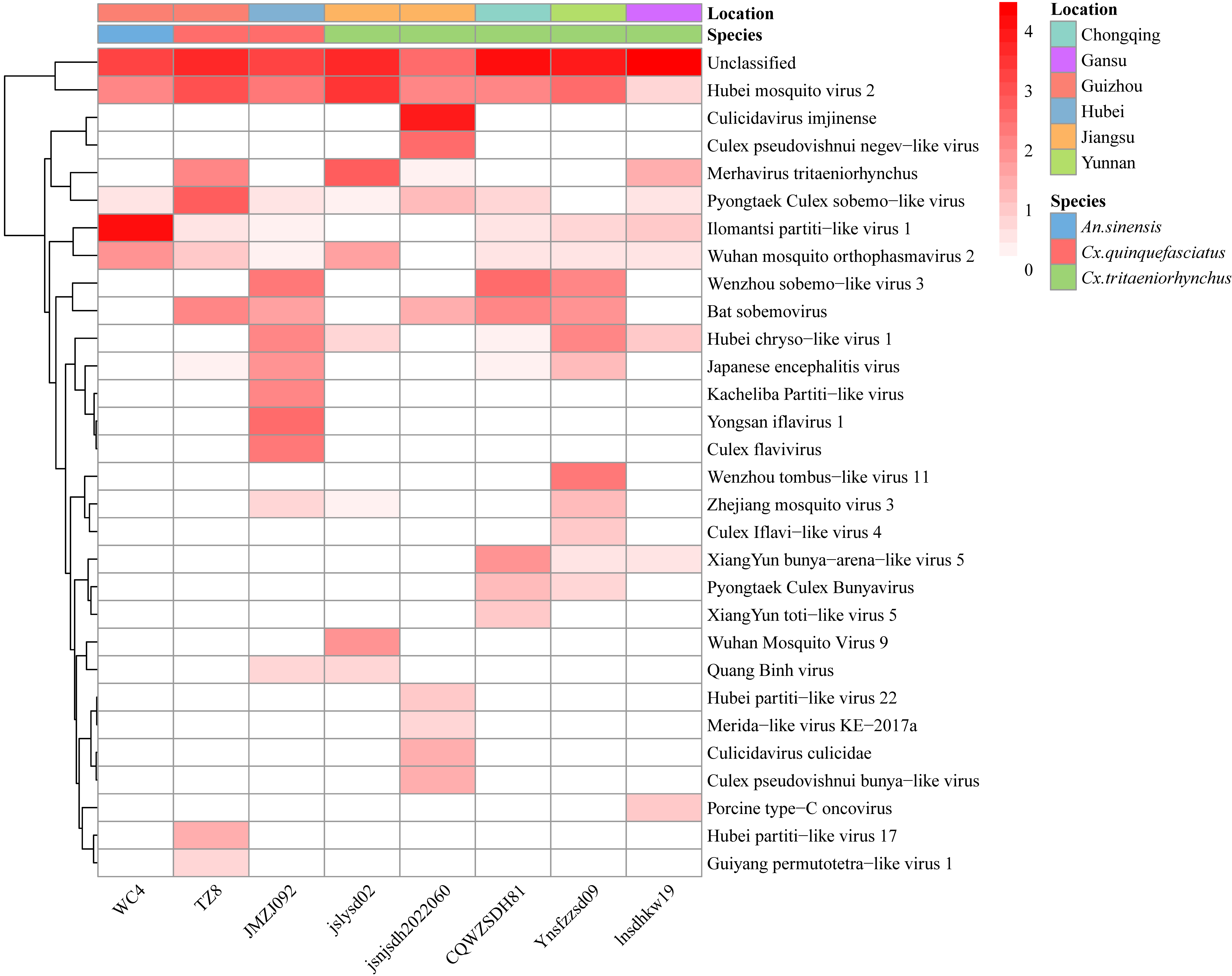
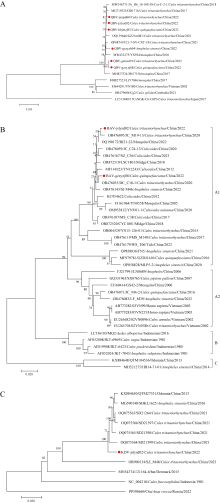
| [1] |
Wang T, Fan ZW, Ji Y, et al. Mapping the distributions of mosquitoes and mosquito-borne arboviruses in China[J]. Viruses, 2022, 14(4):691.
|
| [2] |
Liang GD, Li XL, Gao XY, et al. Arboviruses and their related infections in China: a comprehensive field and laboratory investigation over the last 3 decades[J]. Rev Med Virol, 2018, 28(1):e1959.
|
| [3] |
Chandrasegaran K, Lahondère C, Escobar LE, et al. Linking mosquito ecology, traits, behavior, and disease transmission[J]. Trends Parasitol, 2020, 36(4):393-403.
doi: S1471-4922(20)30021-0
pmid: 32191853
|
| [4] |
de Almeida JP, Aguiar ER, Armache JN, et al. The virome of vector mosquitoes[J]. Curr Opin Virol, 2021, 49:7-12.
doi: 10.1016/j.coviro.2021.04.002
pmid: 33991759
|
| [5] |
闫婷婷, 吴丹, 郑徽, 等. 中国2019—2022年流行性乙型脑炎流行病学特征[J]. 中国疫苗和免疫, 2024, 30(2):160-164.
|
| [6] |
Pan YF, Zhao HL, Gou QY, et al. Metagenomic analysis of individual mosquito viromes reveals the geographical patterns and drivers of viral diversity[J]. Nat Ecol Evol, 2024, 8(5):947-959.
|
| [7] |
Du J, Li F, Han YL, et al. Characterization of viromes within mosquito species in China[J]. Sci China Life Sci, 2020, 63(7):1089-1092.
doi: 10.1007/s11427-019-1583-9
pmid: 31834603
|
| [8] |
Wang YH, Lin XJ, Li C, et al. Metagenomic sequencing reveals viral diversity of mosquitoes from Shandong Province, China[J]. Microbiol Spectr, 2024, 12(4):e0393223.
|
| [9] |
Walker PJ, Freitas-Astúa J, Bejerman N, et al. ICTV virus taxonomy profile: Rhabdoviridae 2022[J]. J Gen Virol, 2022, 103(6):001689.
|
| [10] |
Wu Z, Liu JY, Feng XW, et al. Identification and molecular characteristics of a novel single-stranded RNA virus isolated from Culex tritaeniorhynchus in China[J]. Microbiol Spectr, 2023, 11(4):e0053623.
|
| [11] |
Fang Y, Zhang Y, Zhou ZB, et al. Co-circulation of Aedes flavivirus, Culex flavivirus, and Quang Binh virus in Shanghai, China[J]. Infect Dis Poverty, 2018, 7(1):75.
|
| [12] |
高玉峰, 程晓兰, 丁晔, 等. 中国西南边境地区蚊类携带病原体调查[J]. 中国国境卫生检疫杂志, 2020, 43(2):91-94.
|
| [13] |
吕顺燕, 李楠, 何于雯, 等. 云南省师宗县蚊虫中版纳病毒核酸检测及其序列分析[J]. 云南畜牧兽医, 2020(3):13-16.
|
| [14] |
吴艳琴, 周友华, 周红宁, 等. 中越边境部分地区蚊虫种类及重要虫媒传染病带毒率调查[J]. 中国病原生物学杂志, 2022, 17(2):236-239.
|
| [15] |
Zhang WJ, Li F, Liu AG, et al. Identification and genetic analysis of Kadipiro virus isolated in Shandong province, China[J]. Virol J, 2018, 15(1):64.
doi: 10.1186/s12985-018-0966-y
pmid: 29625620
|
| [16] |
吴云姣, 林小娟, 张晓, 等. 山东省2021年蚊虫中Kadipiro病毒的全基因组序列特征[J]. 病毒学报, 2023, 39(5):1336-1342.
|
| [17] |
殷启凯, 付士红, 王环宇, 等. 我国蚊传虫媒病毒及蚊传虫媒病毒病现状及展望[J]. 中国热带医学, 2024, 24(4):478-485.
doi: 10.13604/j.cnki.46-1064/r.2024.04.21
|
| [18] |
Tang XM, Li RT, Qi YF, et al. The identification and genetic characteristics of quang Binh virus from field-captured Culex tritaeniorhynchus (Diptera: Culicidae) from Guizhou Province, China[J]. Parasit Vectors, 2023, 16(1):318.
|
| [19] |
李樊, 殷启凯, 胡伟军, 等. 我国西北地区蚊虫标本中广平病毒的检测[J]. 疾病监测, 2022, 37(3):373-376.
|
| [20] |
刘红, 梁国栋. 新发虫媒病毒:基因A型版纳病毒全基因组序列扩增引物[J]. 中国媒介生物学及控制杂志, 2016, 27(6):533-538.
doi: 10.11853/j.issn.1003.8280.2016.06.002
|
| [21] |
徐普庭, 王逸民, 左建民, 等. 从云南省无名热病人和脑炎病人分离到新环状病毒[J]. 病毒学报, 1990, 6(1):27-33,100.
|
| [22] |
Li JF, Guo XF, Li YH, et al. Evolutionary analysis of a newly isolated Banna virus strain from Yunnan, China[J]. Arch Virol, 2022, 167(4):1221-1223.
doi: 10.1007/s00705-022-05403-z
pmid: 35277776
|
| [23] |
程睿, 付士红, 范娜, 等. 中华按蚊分离的版纳病毒全基因组序列测定与分子遗传进化分析[J]. 中国媒介生物学及控制杂志, 2018, 29(6):6-12.
|
 ), LI Jinyu2, LUN Xinchang2, LIU Pengbo2, SONG Xiuping2, GUO Yuhong2, LIU Xiaobo2, YUE Yujuan2, LU Liang2, ZHAO Ning2(
), LI Jinyu2, LUN Xinchang2, LIU Pengbo2, SONG Xiuping2, GUO Yuhong2, LIU Xiaobo2, YUE Yujuan2, LU Liang2, ZHAO Ning2(