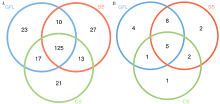
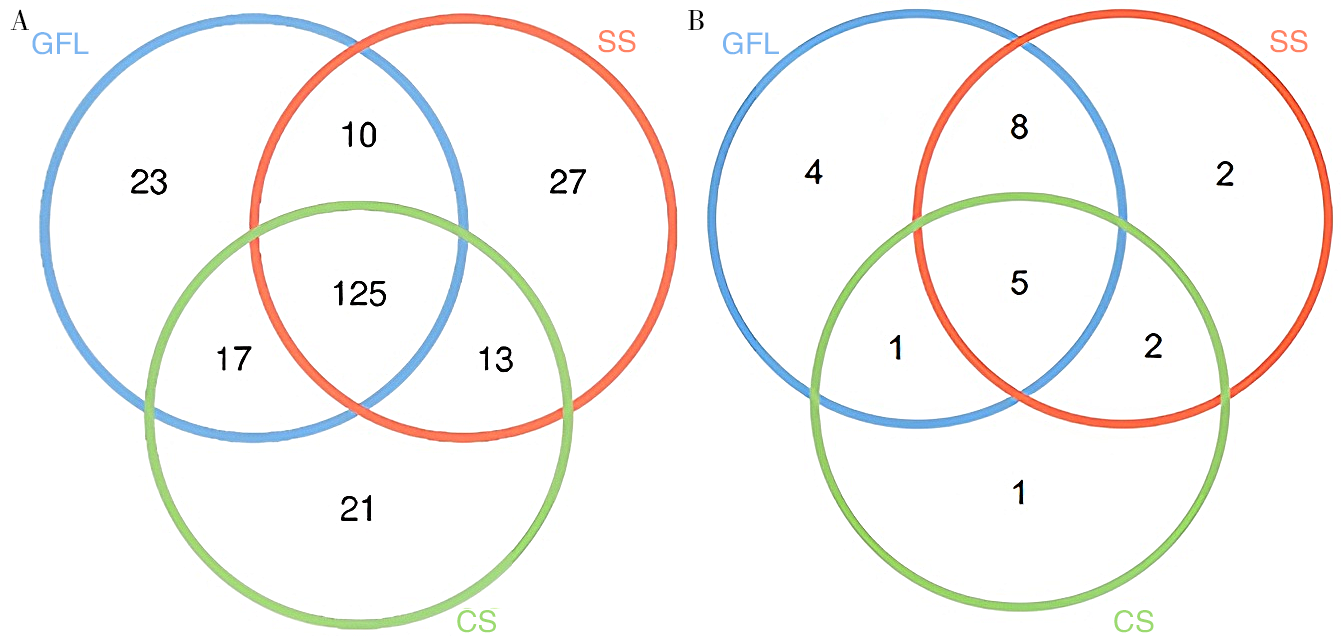
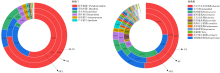
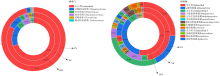
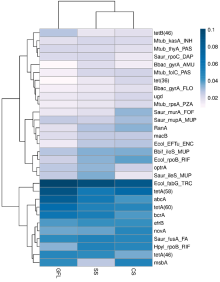
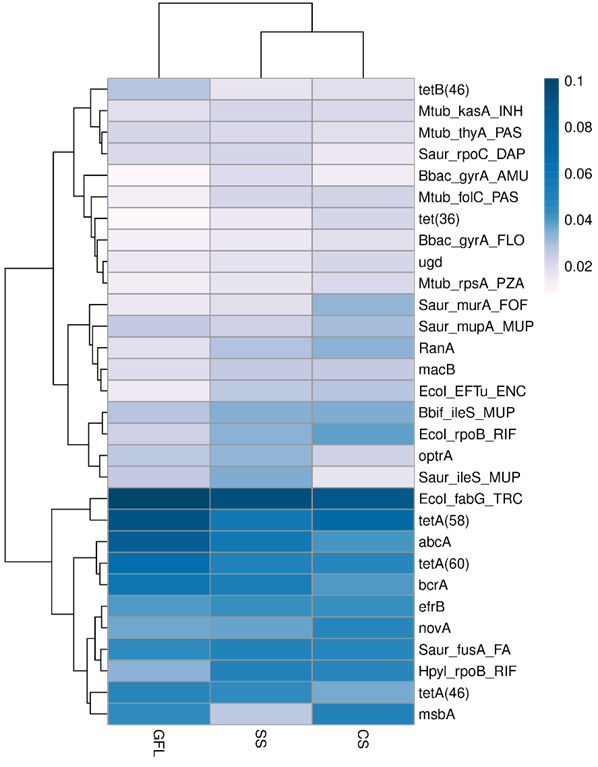
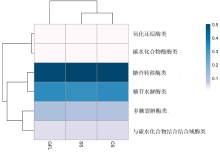
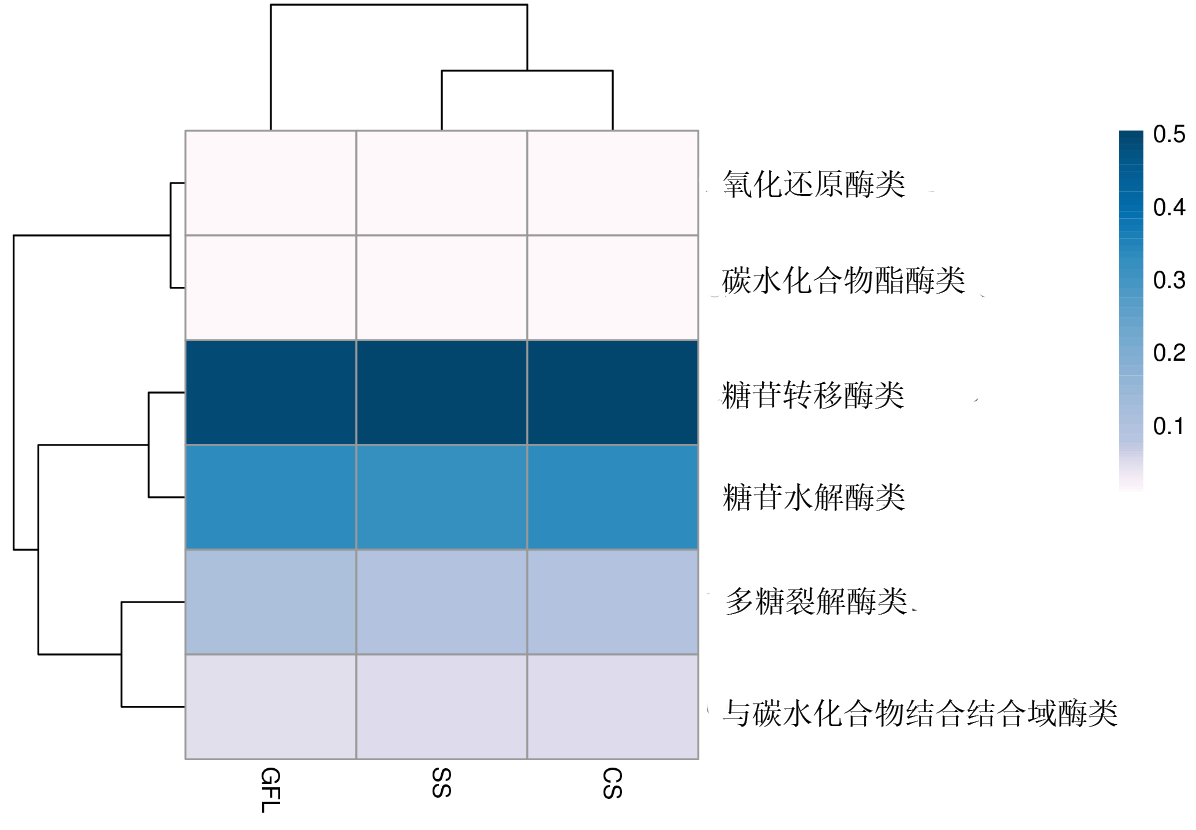
| [1] |
杨舒然, 刘起勇. 白纹伊蚊的全球分布及扩散趋势[J]. 中国媒介生物学及控制杂志, 2013, 24(1):1-4.
|
| [2] |
邓惠. 广东省白纹伊蚊内共生菌Wolbachia的检测及其生理功能研究[D]. 广州: 华南农业大学, 2018.
|
| [3] |
Seabourn P, Spafford H, Yoneishi N, et al. The Aedes albopictus (Diptera: Culicidae) microbiome varies spatially and with ascogregarine infection[J]. PLoS Negl Trop Dis, 2020, 14(8):e0008615.
|
| [4] |
王晓明, 吴焜, 陈晓光, 等. 蚊虫共生微生物群多样性及功能的研究进展[J]. 中国寄生虫学与寄生虫病杂志, 2017, 35(3):305-312.
|
| [5] |
李博琦, 刘铸, 李春晓. 蚊虫共生微生物研究进展[J]. 寄生虫与医学昆虫学报, 2021, 28(4):256-262.
|
| [6] |
Yachida S, Mizutani S, Shiroma H, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer[J]. Nat Med, 2019, 25(6):968-976.
doi: 10.1038/s41591-019-0458-7
pmid: 31171880
|
| [7] |
Saito K, Koido S, Odamaki T, et al. Metagenomic analyses of the gut microbiota associated with colorectal adenoma[J]. PLoS One, 2019, 14(2):e0212406.
|
| [8] |
夏骜, 顾玲, 赵永俏, 等. 白纹伊蚊共生伊丽莎白菌分布特征及对蚊卵孵化和幼虫生长的影响[J]. 热带医学杂志, 2024, 24(8):1109-1114,插页1.
|
| [9] |
He WQ, Chen YX, Zhang XR, et al. Virome in adult Aedes albopictus captured during different seasons in Guangzhou City,China[J]. Parasit Vectors, 2021, 14(1):415.
|
| [10] |
Pan YF, Zhao HL, Gou QY, et al. Metagenomic analysis of individual mosquito viromes reveals the geographical patterns and drivers of viral diversity[J]. Nat Ecol Evol, 2024, 8(5):947-959.
|
| [11] |
Bennett KL, Gómez-Martínez C, Chin Y, et al. Dynamics and diversity of bacteria associated with the disease vectors Aedes aegypti and Aedes albopictus[J]. Sci Rep, 2019, 9(1):12160.
doi: 10.1038/s41598-019-48414-8
pmid: 31434963
|
| [12] |
郭仕辉, 余永涛, 万佳宏, 等. 变形菌门与哺乳动物结肠肠道菌群失调相关研究进展[J]. 中国微生态学杂志, 2022, 34(4):479-484.
|
| [13] |
樊洁丽, 刘焱晖, 殷雅楠, 等. 基于宏基因组学分析海南省不同养殖场库蠓共生微生物群落组成[J]. 中国媒介生物学及控制杂志, 2023, 34(4):472-479.
|
| [14] |
Jordan CKI, Brown RL, Larkinson MLY, et al. Symbiotic Firmicutes establish mutualism with the host via innate tolerance and resistance to control systemic immunity[J]. Cell Host Microbe, 2023, 31(9):1433-1449. e9.
doi: 10.1016/j.chom.2023.07.008
pmid: 37582375
|
| [15] |
Zheng XY, Zhang DJ, Li YJ, et al. Incompatible and sterile insect techniques combined eliminate mosquitoes[J]. Nature, 2019, 572(7767):56-61.
|
| [16] |
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue,Chikungunya,and Plasmodium[J]. Cell, 2009, 139(7):1268-1278.
|
| [17] |
郑小英, 吴瑜, 张东京, 等. 沃尔巴克氏体(Wolbachia)结合昆虫绝育技术控制白纹伊蚊种群[J]. 南京农业大学学报, 2020, 43(3):387-391.
|
| [18] |
李辞修. 节肢动物中负链RNA病毒的多样性及其进化[D]. 北京: 中国疾病预防控制中心, 2015.
|
| [19] |
Petersen JM, Bézier A, Drezen JM, et al. The naked truth:an updated review on nudiviruses and their relationship to bracoviruses and baculoviruses[J]. J Invertebr Pathol, 2022, 189:107718.
|
| [20] |
秦翊翔, 蓝如束, 覃慧芳, 等. 广西地区结核分枝杆菌异烟肼耐药基因突变特征分析[J]. 中国感染控制杂志, 2023, 22(3):280-286.
|
| [21] |
邬俊勇, 黄韬, 江光荣. 2019—2021年宜春地区患者结核分枝杆菌利福平耐药株基因型分析研究[J]. 中国医学创新, 2024, 21(13):117-120.
|
| [22] |
Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013[J]. Nucleic Acids Res, 2014, 42(Database issue):D490-D495.
|
| [23] |
武佳慧, 宋晓, 彭荟, 等. 小核糖核酸调控蚊虫代谢解毒酶基因表达的研究进展[J]. 中国热带医学, 2023, 23(7):766-772.
doi: 10.13604/j.cnki.46-1064/r.2023.07.16
|
 ), 张驰1, 庞博文1, 吴佳玲1, 蒋露芳2, 吕锡宏1(
), 张驰1, 庞博文1, 吴佳玲1, 蒋露芳2, 吕锡宏1(